Channels
Special Offers & Promotions
Machine Intelligence for designing molecules and reaction pathways
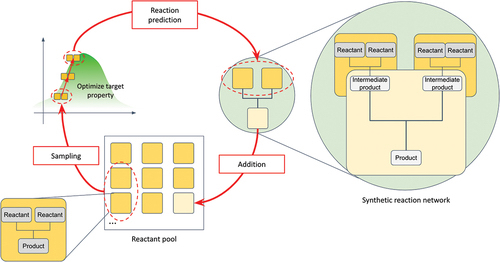
Two key challenges in chemistry innovation are solved simultaneously by exploring chemical opportunities with artificial intelligence.
Researchers in Japan have developed a machine learning process that simultaneously designs new molecules and suggests the chemical reactions to make them. The team, at the Institute of Statistical Mathematics (ISM) in Tokyo, published their results in the journal Science and Technology of Advanced Materials: Methods.
Many research groups are making significant progress in using artificial intelligence (AI) and machine learning methods to design feasible molecular structures with desired properties, but progress in putting the design concepts into practice has been slow. The greatest impediment has been the technical difficulties in finding chemical reactions that can make the designed molecules with efficiencies and costs that could be practicable for real-world uses.
“Our novel machine learning algorithm and associated software system can design molecules with any desired properties and suggest synthetic routes for making them from an extensive list of commercially available compounds,” says statistical mathematician Ryo Yoshida, leader of the research group.
The process uses a statistical approach called Bayesian inference which works with a vast set of data about different options for starting materials and reaction pathways. The possible starting materials are all combinations of the millions of compounds that can be readily purchased. The computer algorithm assesses the huge range of feasible reactions and reaction networks to discover a synthetic route towards a compound with the properties it has been instructed to aim for. Expert chemists can then review the results to test and refine what the AI proposes. AI makes the suggestions while humans decide which is best.
“In a case study for designing drug-like molecules, the method showed overwhelming performance,” says Yoshida. It also designed routes towards industrially useful lubricant molecules.
“We hope that our work will accelerate the process of data-driven discovery of a wide range of new materials,” Yoshida concludes. In support of this aim, the ISM team has made the software implementing their machine learning system available to all researchers on the GitHub website.
The current success focused only on the design of small molecules. The team now plan to investigate adapting the procedure to design polymers. Many of the most important industrial and biological compounds are polymers, but it has proved difficult to make new versions proposed by machine learning due to challenges in finding reactions to build the designs. The simultaneous design and reaction discovery options offered by this new technology might break through that barrier.
About Science and Technology of Advanced Materials: Methods (STAM-M)
STAM Methods is an open access sister journal of Science and Technology of Advanced Materials (STAM), and focuses on emergent methods and tools for improving and/or accelerating materials developments, such as methodology, apparatus, instrumentation, modeling, high-through put data collection, materials/process informatics, databases, and programming.
News Channels
- Latest News
- New Laboratory Products
- Industry News
- Laboratory Automation | IT Solutions
- Microscopy | Image Analysis
- Separation Science
- Research | Case Studies
- Video Presentations
- Events | Conferences
Subscribe to any of our newsletters for the latest on new laboratory products, industry news, case studies and much more!

Popular this Month
Top 10 most popular articles this month
Today's Picks
Looking for a Supplier?
Search by company or by product
Please note Lab Bulletin does not sell, supply any of the products featured on this website. If you have an enquiry, please use the contact form below the article or company profile and we will send your request to the supplier so that they can contact you directly.
Lab Bulletin is published by newleaf marketing communications ltd.
Media Partners